ΤΙ ΕΙΝΑΙ ΣΠΑΝΙΑ ΝΟΣΟΣ
Σύμφωνα με τον ορισμό της Ευρωπαϊκής Ένωσης (ΕΕ) για τη δημόσια υγεία, σπάνια χαρακτηρίζεται κάθε νόσος που προσβάλλει λιγότερα από 5 άτομα στα 10.000. Οι σπάνιες παθήσεις πλήττουν περίπου το 8% των ανθρώπων, έχουν πιστοποιηθεί και ανέρχονται σε 6 – 8.000. Οι πιο πολλές από αυτές είναι χρόνιες, εξελισσόμενες, μπορούν να προκαλέσουν αναπηρία, και αρκετές από αυτές είναι απειλητικές για τη ζωή. Στη χώρα μας υπάρχουν περίπου 880.000-1.00.000 ασθενείς με σπάνια νοσήματα. Για τα περισσότερα δεν υπάρχει θεραπεία ή ίαση. Το 80% αυτών έχουν γενετική προέλευση, ενώ το 75% εμφανίζεται στην παιδική ή εφηβική ηλικία.
ΤΙ ΕΙΝΑΙ ΛΥΣΟΣΩΜΙΚΑ ΑΘΡΟΙΣΤΙΚΑ ΝΟΣΗΜΑΤΑ
Λυσοσωμικά αθροιστικά νοσήματα
Τα λυσοσωμικά αθροιστικά νοσήματα (Lysosomal Strorage Diosordes LSD’s) ειναι μια ομάδα 50 σπάνιων κληρονομικών μεταβολικών νοσημάτων, με συνολική επίπτωση 1:700 γεννήσεις, που οφείλονται σε έλλειψη ενζύμων (πρωτεϊνών) από τα κύτταρα και οδηγούν σε σύνθετες διαταραχές του μεταβολισμού. Συνήθως παρουσιάζουν εκδηλώσεις σε πολλά συστήματα οργάνων, συμπεριλαμβανομένου του σκελετού, του εγκεφάλου, του δέρματος, της καρδιάς και του κεντρικού νευρικού συστήματος. Εξαιτίας της πολυπλοκότητας των συμπτωμάτων πολλοί ασθενείς ταλαιπωρούνται μήνες ή και χρόνια μέχρι να ταυτοποιηθεί και να τεθεί η τελική διάγνωση.

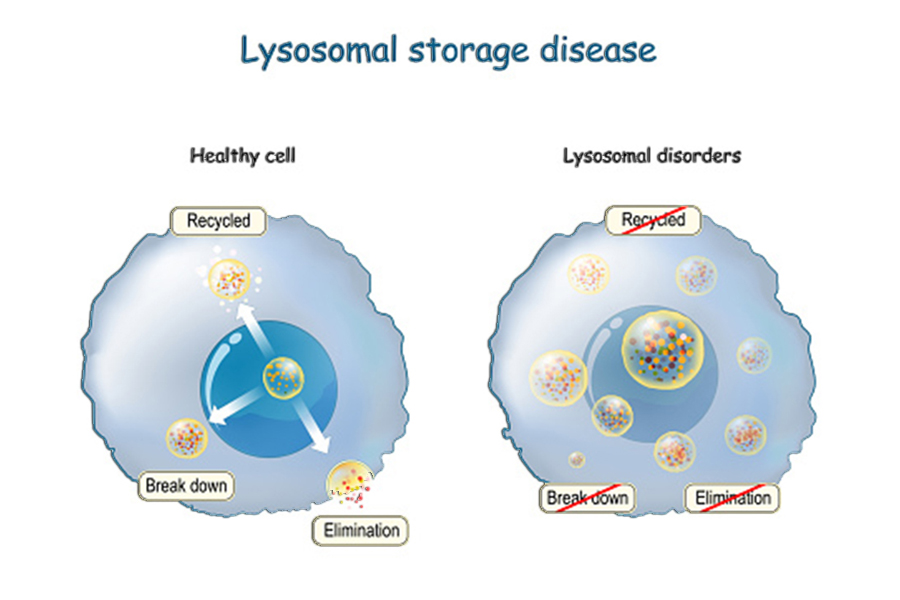
Ένας τρόπος κατηγοριοποίησης είναι ο παρακάτω:
- Σφιγγολιπιδώσεις: νόσος Niemann-Pick τύπου Α, νόσος Gaucher (τύποι I, II και III), ασθένεια Krabbe, νόσος Fabry, γαγγλιοσιδώσεις GM1 και GM2, ασθένεια Morquio Β, μεταχρωματική λευκοδυστροφία, ασθένεια Farber
- Νόσοι αποθήκευσης γλυκογόνου τύπου II: νόσος Pompe
- Βλεννοπολυσακχαριδώσεις ή σύνδρομα MPS : σύνδρομα Hurler, Hurler-Scheie, Scheie, Hunter, Sanfilippo, Morquio, Maroteaux-Lamy και Sly
- Ολιγοσακχαριδώσεις: ασθένειες Schindler και Kanzaki, α- και β-μαννοσίδωση, α-φουκοσίδωση, σιαλίδωση, γλυκοζουρία
- Λιπιδώσεις: νόσος Niemann-Pick, ασθένεια Wolman
Περισσότερο αναλύονται οι εξής: